受体酪氨酸激酶c-MET(以下简称MET)的功能失调已被证实是肿瘤发生的驱动因素。与许多其他原癌基因相比,MET的显著特点是三种不同类型的基因突变均可导致肿瘤的发生,包括扩增、突变和融合等。随着MET驱动突变癌症的诊断和治疗的迅速发展,《自然评论临床肿瘤学》(Nature Reviews Clinical Oncology)发表一篇关于MET基因的综述文章,系统阐述了MET驱动突变癌症的临床特征、诊断策略和靶向治疗方案1。

一、MET扩增
MET扩增即MET拷贝数扩增,包括整体染色体重复和局部区域基因的重复整体染色体重复即多倍体(polysomy)肿瘤细胞中出现多条7号染色体,多倍体在生物学中不能作为驱动基因。扩增是局部或区域基因的重复,断裂-融合-桥连机制是导致基因扩增的主要原因。与多倍体比较,MET扩增可能作为驱动基因,也是EGFR-TKI耐药的主要机制之一。MET基因拷贝数为连续变量,阳性阈值的定义会影响发生率、与其他基因亚型重叠率,及作为MET抑制剂疗效预测指标的能力。如下介绍MET扩增的诊断、临床特征和治疗等。
| ET扩增检测
MET扩增可使用多种技术检测,包括荧光原位杂交(FISH)、实时定量PCR(qRT PCR)和二代测序(NGS)(见图1)。
荧光原位杂交(FISH)
FISH是一种常用的检测技术,其基本原理是利用荧光基团偶联标记的DNA探针来识别特定序列的基因组区域,常用于福尔马林固定石蜡包埋(FFPE)组织切片标本的检测。当待探测的基因组序列暴露于荧光标记的探针后,会发出相应颜色的荧光。荧光信号的数量则代表了了基因的拷贝数。
FISH主要有两种主要方式来检测MET扩增。第一种方法直接测定MET基因拷贝数(GCN)。目前临床研究采用最多的是Cappuzzo标准,即平均每个细胞有5条及其以上MET基因拷贝数(MET GCN ≥5)可视为MET扩增,也有其他学者建议平均拷贝数≥6个甚至15个才能定义为MET扩增。遗憾的是这种通过拷贝数来确定扩增的方法并不能区分是多倍体还是局部扩增。第二种方法将MET/CEP7≥2.0作为MET扩增标准,这是目前最广为接受的标准。也有学者根据MET/CEP7值将扩增程度分为三组:低度扩增(1.8 - 2.2)、中度扩增(2.2 -5)和高度扩增组(≥5)。
二代测序(NGS)
与适用于FISH的标准相似,对于MET扩增的单一定义没有达成共识。目前大部分平台均使用读取深度(read depth)测定法,这种方法基于读长深度的信号与样本中染色体片段的拷贝数成比例的原理,并使用一些生物信息学方法进行计算,然而在敏感性和特异性上还是有一定局限。有的平台还会使用同一患者来源的良性组织或者血液样本作为对照以进一步提高敏感度,NGS的优势在于除了能够同时评估数百个基因的变异外,还具有高分辨率和从众多的复制染色体中识别局部基因扩增的能力。临床上有两种基于NGS的检测方法:基于扩增子的NGS和基于捕获的NGS技术(见图1a、1b)。
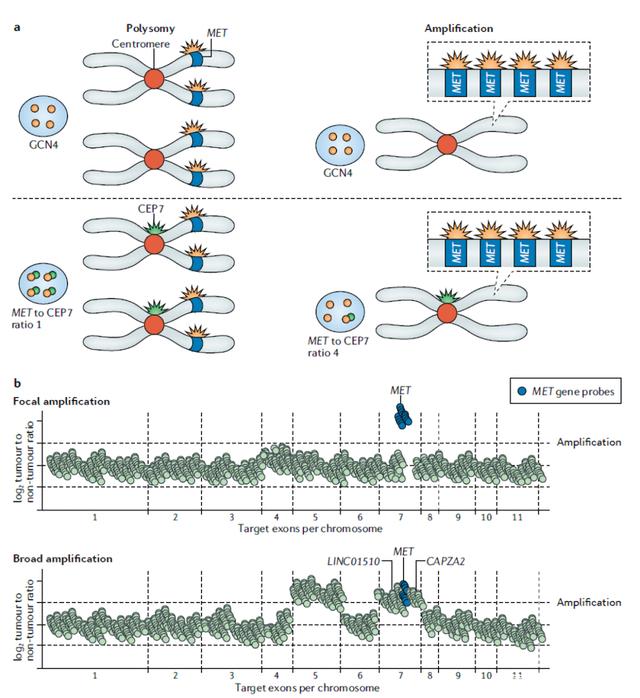
图1.MET扩增肿瘤诊断
| 临床病理特征
原发MET扩增
在多种实体瘤中都发现有原发MET扩增,根据肿瘤基因组计划(TCGA)和cBioPortal数据库提供的数据,MET扩增比较典型的有非小细胞肺癌(NSCLC) ,检出率约<1-5%;胃癌中约<1-10%;大约2-4%的结肠癌(CRCs),13%左右的I型肾乳头状癌(PRCCs)和3%左右的II型肾乳头状癌也检出MET扩增,在食管癌和肝细胞癌中检出比例较低。在许多恶性肿瘤中MET扩增意味着预后不良。研究发现NSCLC细胞MET扩增度越高,肿瘤的MET驱动依赖性越强。MET高度扩增 (MET /CEP7 ≥5,FISH法)的 NSCLC患者,很少合并其他致癌基因的驱动改变(比如EGFR突变或者ALK融合),而在MET低度扩增(MET / CEP7 ≥1.8 , ≤2.2,FISH法)和中度扩增 (MET / CEP7>2.2 ,<5,FISH法)的患者中,更多合并有其他驱动基因改变(MET高,中,低度扩增合并其他驱动基因改变比例分别为0%,52%,50%)。
获得性MET扩增
MET扩增可以是原发的也可以是被其他原癌驱动基因(EGFR)次级激活,这也是EGFR等TKI耐药机制之一。根据不同的检测方法和平台提供的数据,在接受一到三代EGFR TKI靶向治疗后的NSCLC患者中大约5%~20%的比例会出现MET扩增。EGFR突变的肿瘤细胞可以绕开EGFR TKI(吉非替尼或者厄洛替尼等)作用的PI3K通路,激活MET这一旁路信号通路,而PI3K通路也可以通过MET介导的HER3信号通路进一步激活。MET扩增也被发现是ALK融合阳性NSCLC癌患者对ALK抑制剂耐药的机制之一。此外,在接受抗EGFR单克隆抗体治疗的结肠癌患者和接受EGFR和BRAF抑制剂联合治疗的BRAF V600E突变的结肠癌患者中也可检测到MET扩增。当患者接受EGFR TKI治疗后MET扩增的次级激活即可发生,这种激活也可以发生在肿瘤细胞的的亚克隆群中。当使用NGS检测的样本混合有MET扩增和非MET扩增的肿瘤细胞时该结果可能会低估MET的扩增程度,为了进一步提高准确度,建议可以联合FISH或者单细胞测序进行补充。
| 靶向治疗
MET扩增水平和MET抑制剂疗效呈正相关。
1)原发性MET扩增
PROFILE 1001研究是最早开展的关于MET抑制剂疗效和MET扩增程度相关性的研究。患者分别分为低度扩增组(MET / CEP7 ≥1.8 , ≤2.2),中度扩增组 (MET / CEP7>2.2 ,<5)和MET高度扩增组 (MET /CEP7 ≥5),结果提示高度扩增组的客观反应率(ORR)可达67%,低扩增组和中扩增组的客观反应率ORR分别为0%和17%。随后研究组将中度扩增组界定值调整为MET /CEP7值为2.2-4.0,高扩增组界定为MET /CEP7 ≥4.0,最终结果提示总体临床获益最显著的也是高度扩增组,其ORR为40%,中位无进展生存期(PFS)为6.7个月,而低度扩增组和中度扩增组的ORR分别为33%和14%,PFS分别为1.8个月和1.9个月(图2)。
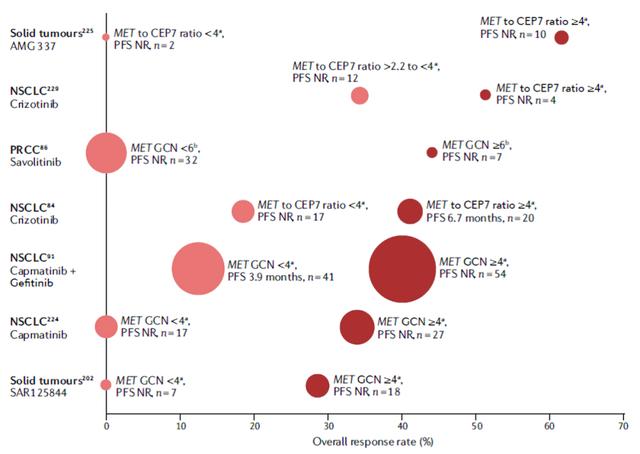
图2.MET扩增肿瘤患者的靶向治疗及其有效性
2)获得性耐药
对于依赖于另一驱动基因且出现MET次级激活的肿瘤患者,针对原发驱动基因和MET信号通路的联合治疗可能有效。例如EGFR突变的患者在使用EGFR TKI后耐药进展,如果是MET扩增导致的耐药,那么联合使用EGFR TKI和MET抑制剂可能是行之有效的方案。研究揭示对于EGFR突变伴随MET扩增耐药(FISH或者NGS检测结果为MET /CEP7≥2或者GCN≥5)的患者使用奥西替尼和沃利替尼联合治疗的ORR可达30%。另一项研究提示接受一/二代EGFR TKI治疗的患者出现MET扩增耐药,接受吉非替尼和特泊替尼联合治疗,其ORR可高达67%。其他药物比如EGFR和 MET双特异性抗体JNJ-372,在EGFR耐药进展的患者中也表现出了良好的活性,这些药物在继发MET扩增的恶性肿瘤中的疗效会有更多研究揭晓。
总而言之,高MET扩增水平预示着使用MET抑制剂会有更好的疗效,这种预测模式对于MET抑制剂单药使用或者MET与其他TKI联合治疗都有预测意义。因此我们再次呼吁尽快实现对MET扩增的标准化定义,不仅仅是为了达到诊断意义,也是为了尽可能识别出MET改变驱动的恶性肿瘤患者并区分其扩增程度,让其从针对MET的靶向治疗获益。
MET突变
激活突变可发生在MET基因不同位置,包括激酶结构域、外显子14侧翼的内含子剪接位点和细胞外结构域(见图3)。
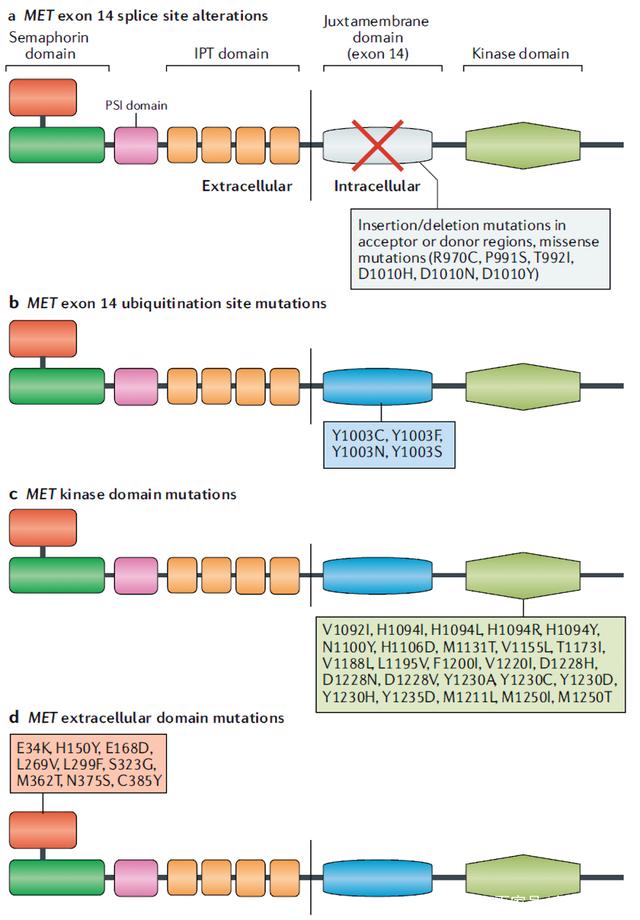
图3.MET突变
激酶结构域突变
1997年首次在遗传性乳头状肾细胞癌(PRCC)患者中发现MET突变,这些胚系突变包含V1092I、 H1094R/Y、M1131T、V1188L、V1220I、M1250T和D1228H/N/V。MET激酶结构域突变增加激酶活性,导致表型转化或肿瘤灶的形成。除了原发性突变,MET激酶活性区也能发生EGFR TKI耐药的继发性突变,在非小细胞肺癌中,研究发现MET外显子14、METY1230C、METY1230H、METD1228H 和METD1228N均能介导克唑替尼的耐药。
MET 14外显子突变
MET激活导致MET激酶活化环内由外显子14编码的近膜结构域的Y1003残基磷酸化。该残基的磷酸化介导MET与c-Cbl E3连接酶结合,导致泛素化并最终降解MET,作为自我调节负反馈环的一部分。在这个过程中,所有的外显子突变都会受到持续的干扰。
| MET突变检测
DNA测序:MET外显子14跳跃突变具有高度的异质性,因此,一个有效的NGS分析必须能够捕捉到这种广泛多样的突变。如前所述,NGS法分为扩增法和杂交捕获法,扩增法使用引物来检测基因组区域目的基因序列,与杂交捕获法相比,这种方法可以缩短检测时间,对一些难以测序的区域有更好的捕获,但对于有重复、偏倚和等位基因丢失等区域容易出现测序失真。一些MET EX14突变比如碱基插入/缺失突变,通常位于扩增区域之外,检测可能会被遗漏。此外碱基插入/缺失突变可能会干扰引物和位点的结合,进而干扰基于引物的扩增法对其进行检测。不少研究表明相当大比例的MET EX14(50%的比例)使用扩增法NGS可能出现假阴性结果。
RNA 测序:DNA测序只能通过检测外显子突变来预测MET外显子跳跃突变,突变发生在转录后的剪接。因此RNA测序弥补DNA测序的不足。然而,RNA的稳定性不如DNA,这限制了组织的保存期限。此外,鉴于mRNA表达在非恶性组织和肿瘤组织中高度变异性,对检测结果的解读带来挑战。目前是RNA测序作为传统测序的辅助手段,如确认MET外显子14跳跃。综上所述,虽然MET 14外显子表达的突变可能难以用基于DNA测序平台来检测,但可以通过RNA测序来直接确定14号外显子是否被转录。
| 靶向治疗
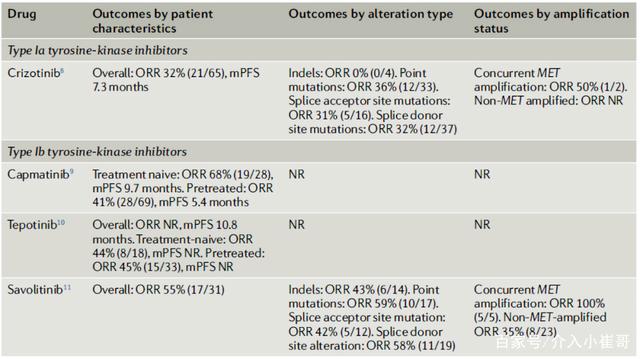
激酶结构域突变:MET靶向治疗对某些MET激酶结构域突变是有效的。然而,各种MET-TKIs对特定突变的活性是有差异的。具体地说,II型MET抑制剂,如卡博替尼和foretinib,对几种激酶结构域突变(如D1228N、M1250T和H1094Y/L145)具有临床前活性,而I型MET抑制剂,如克唑替尼,对引起这些突变的肿瘤缺乏任何实质性活性。因此,MET激酶结构域突变已成为MET扩增和MET外显子14跳跃突变的癌症患者对克唑替尼的耐药机制。
MET外显子14跳跃突变:与能够改变MET激酶构象的激酶结构域突变相反,MET外显子14跳跃突变理论上同样均有与野生型MET相似的激酶结构域。Ⅰ型和Ⅱ型MET-TKIs都能抑制这种变异体,而且这两种类型在MET外显子14跳跃突变的癌症模型中都显示了临床前活性。前瞻性的临床研究证实克挫替尼(Ia型MET抑制剂)针对MET外显子14跳跃突变的NSCLC有效(ORR 32%),并且Ib型MET抑制剂卡马替尼、特泊替尼和沃利替尼也对MET外显子14跳跃突变有很好的疗效(见表1)。
表1. MET外显子14跳跃突变肺癌的靶向治疗
MET融合
MET是最初在化学转化骨肉瘤细胞系中发现含有TPR–MET融合后被鉴定为癌基因。随后在胃癌、甲状腺癌、PRCC、肺腺癌、肝癌、胶质瘤和肉瘤患者中发现MET融合。融合的确切频率尚不明确,尽管它们在胶质瘤中丰富,发生在约12%的患者。除TPR-MET外,还发现了多个其他MET融合(见图4)。这些融合可以通过染色体内融合(如PTPRZ1–MET、CLIP2–MET、CAPZA2–MET和ST7–MET)或染色体间融合(如KIF5B–MET,TPR–MET,GPRC5C–MET和CD47–MET),每种类型约占所有MET融合的一半。在患有胶质母细胞瘤的儿科患者中,染色体内融合似乎更为常见最常见的是PTPRZ1。
MET融合通常包括该基因的15外显子,该外显子编码激酶域,而许多上游伴侣基因编码二聚体结构域,导致配体无关的连接性MET激活。此外,一些融合事件(如TPR–MET)被发现排除了外显子14,从而使得MET激活机制类似于MET外显子14跳过的机制。有趣的是,包含外显子14的融合(如KIF5B-MET和PTPRZ-MET)似乎比排除外显子14的融合更不易致癌。在PTPRZ-MET融合中,PTPRZ启动子通常与全长MET基因融合,包括外显子2上的MET二聚结构域,这种融合导致MET过表达和下游信号的激活增加。
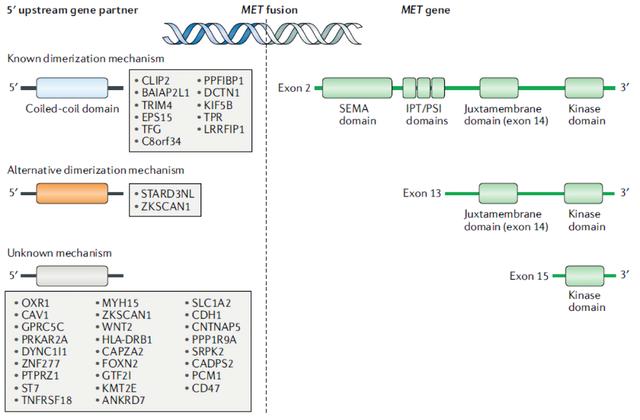
图4.MET基因融合
| MET融合检测
多种分析技术可用于检测MET融合,包括FISH、RT-PCR和NGS。然而,这些平台都没有在这一应用中得到了很好的研究,而复杂的和/或新的MET融合和重排特别难用FISH来检测。因此目前越来越关注NGS,这是临床上检测基因变异的首选方法。基于DNA的NGS能够可靠地检测各种MET融合。虽然DNA-NGS检测基因融合也存在着局限性,如融合断点发生在重复的内含子区域;内含子区域太长,不易设计引物;检测新的融合基因伴侣也有限。正与前面提到的,这些不足之处都可以通过RNA-NGS检测来避免。在一项研究中,DNA-NGS检测为驱动基因阴性的NSCLC队列,用RNA-NGS检测,发现12%的患者存在驱动基因融合2。这一研究结果表明,在MET融合基因检测上,RNA-NGS可以补充基于DNA的NGS检测,特别是在无驱动基因突变的情况下。
| 靶向治疗
迄今为止,MET靶向治疗在MET融合阳性癌症患者中的作用几乎被忽视。MET TKIs在TPR-MET转化细胞系中能够诱导细胞凋亡,在MET融合阳性的肺腺癌或神经胶质瘤病例中,已经报道了克唑替尼有效。目前正在进行多项旨在评估MET TKIs疗效的临床试验,包括对伴有MET融合的肿瘤患者的试验(NCT02978261, NCT03993873和NCT01639508)。然而,MET TKI的疗效在一个大的同质的MET融合阳性的队列中尚未被报道。
MET过表达
MET可在缺氧和/或炎症条件下转录诱导癌细胞增殖,减少凋亡,促进转移。因此,即使在缺乏诸如MET扩增、突变或融合等基因组驱动因素的情况下,肿瘤也可能依赖MET信号。关于MET过表达的诊断和治疗如下:
| MET过表达检测
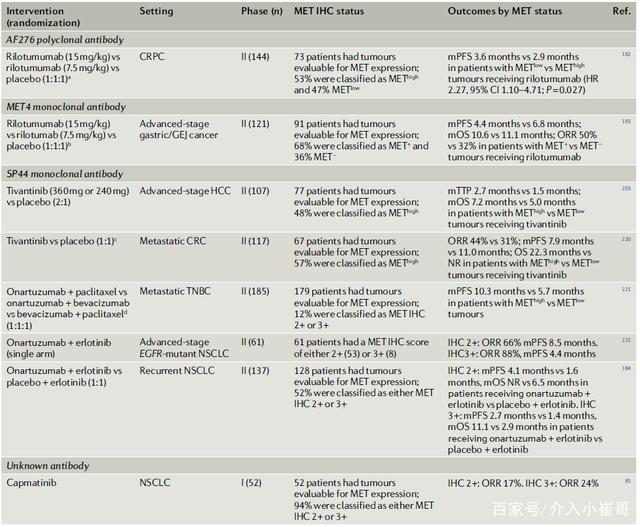
免疫组织(IHC):许多抗MET抗体已被用于免疫组化(IHC)检测。这些包括单克隆抗体(如SP44、cMET和MET4)、多克隆抗体(如多克隆MET AF276)和磷酸化MET抗体(如pMET Y1349)。病理学家评估的IHC染色的程度和强度提供了MET蛋白表达半定量指标。IHC使用各种评分系统来定义MET表达和过表达。
质谱法(Mass spectrometry):选择反应监测质谱法(SRM-MS)能在福尔马林固定的石蜡包埋组织切片中定量测定MET,结果重现性好,即使在固定时间≥1年的样本中也是如此。与IHC相比,SRM-MS不易受到观察者间偏差的影响,并且可以检测较低水平的蛋白质表达。SRM-MS不能区分肿瘤和非恶性组织中表达的蛋白质,因此MET定量可能受到混合基质和/或炎症浸润的影响。此外,SRM-MS比IHC技术要求更高,成本也更高,这意味着目前这种方法的应用不太广泛。IHC目前通常用于诊断病理学实验室,而SRM-MS的使用在很大程度上仍处于研究阶段。
| 靶向治疗
目前,MET-TKIs在MET过表达的肿瘤患者中几乎没有活性(表2),庆幸的是,新的MET靶向治疗策略正在开发,如双特异性抗体,抗体组合和ADC等。
表2. 根据MET表达状态进行靶向治疗的结果
结论
鉴定MET驱动突变癌症患者的过程很复杂。从诊断的角度来看,需要对连续变量(包括MET扩增水平或MET表达水平)标准化成具有临床意义的阈值,然后这些特征才能用来指导治疗相关的决策。为了最大限度地提高检测MET扩增、突变和/或融合的可能性,可能需要将诊断学转向使用更全面和技术复杂的检测方法。应该考虑用NGS方法同时检测肿瘤活检和血浆样本中DNA和RNA水平的变异。鉴于MET靶向治疗在许多此类癌症中非常有效,因此有效检测MET驱动突变癌症至关重要。
参考资料:
1.Robin Guo, Jia Luo, Jason Chang,et al.MET-dependent solid tumours — molecular diagnosis and targeted therapy.Nature Reviews Clinical Oncology volume 17, pages 569–587(2020).
2.Benayed, R. et al. High yield of RNA sequencing fortargetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden.Clin. Cancer Res. 25, 4712–4722 (2019).